ANEMIA HEMOLÍTICA.

Representan el
5% de todas las anemias y pueden ocurrir en personas de cualquier edad con un
índice de mortalidad en general bajo. Se producen por un aumento en la
destrucción de los eritrocitos con una disminución en su supervivencia a menos
de 100 días.
Aunque la hemólisis puede cursar
asintomática, lo más común es que se desarrolle anemia cuando la destrucción
celular sobrepasa las capacidades de la eritropoyesis.
Hay dos mecanismos por los cuales
se `produce destrucción de los eritrocitos:
intravascular o extracorpuscular y extravascular o intracorpuscular,
siendo esta última más común.
MANIFESTACIONES CLINICAS.
Una sintomatología general para
cualquier anemia hemolítica:
·
Ictericia
·
Subictericia
·
Esplenomegalia
·
Fiebre
·
Dolor abdominal
·
Hemosiderosis
·
Litiasis biliar
·
Ulceraciones en las extremidades.
DIAGNÓSTICO.
Se dará por el síndrome hemolítico,
exploración y valores analíticos (la hemoglobina puede oscilar entre 5 a 9 g/dl
y los valores de hematocrito 15% y 30%).
TRATAMIENTO.
En ambos tipos se dará un
tratamiento general de los síndromes talasémicos que consistirá en un consejo
genético y un diagnóstico prenatal, además del tratamiento con ácido
fólico. Solo en el caso de la talasemia
mayor harán falta transfusiones e incluso intervenciones como esplenectomía y/o
trasplante de médula ósea.
ANEMIA FERROPENICA O POR
DEFICIENCIA DE HIERRRO.Se caracteriza por un descenso de
los valores de hierro en el organismo, no llevándose a cabo una eritropoyesis
eficaz. En el caso de la embarazada, esta se puede producir por una dieta
deficitaria y/o aumento de los requerimientos.
MANIFESTACIONES CLINCIAS.
Astenia progresiva.
Palidez mucocutánea
Acúfenos
Sensación de mareos, vértigos.
Intolerancia a la actividad
Palpitaciones
Disnea de esfuerzo.
Dolor pericordial en casos severos.
Glositis
·
Cabello frágil y uñas quebradizas.
·
Sensación de quemazón y prurito
·
Alteración del sentido del gusto
·
Susceptibilidad e irritación.
DIAGNOSTICO.
Además del síndrome anémico
tendrá que confirmarse el tipo con valores analíticos por lo que cuando el
valor hematocrito es menor al 32% y cuando la hemoglobina desciende por debajo
de 11.5 g/dl.
TRATAMIENTO.
Primordial el aumento de hierro
en la dieta, aplicar refuerzo farmacológico
por vía oral o intramuscular.
ANEMIA MEGALOBLASTICA.
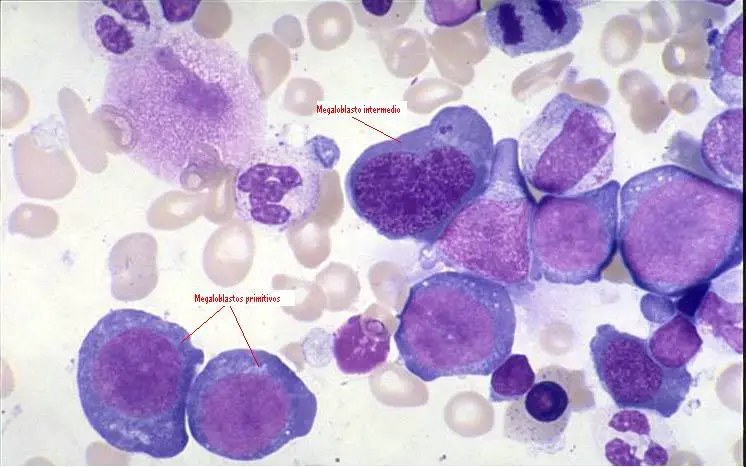
Se produce por la alteración en
la formación de hematíes a causa del déficit de elementos maduradores de los
eritrocitos. Esto es debido a un déficit en los valores de ácido fólico y
vitamina B12 ya sea por una disminución en la ingesta o aumento en los
requerimientos debido al embarazo o la lactancia.
MANIFESTACIONES CLINICAS.
Se dará el síndrome anémico y de los síntomas
que lo conforman además de la palidez puede aparecer subictericia.
TRATAMIENTO.
El déficit de vitamina B12 y ácido fólico
además de con dieta se suplirá con tratamiento famacológico por vía oral.
ANEMIA DE CÉLULAS FALCIFORMES
Es un tipo de anomalía genética
que afecta a la producción, estructura y funcionamiento de la hemoglobina. Esta
hemoglobinopatía consiste en que la persona presenta como rasgo principal HbS
en vez de HbA que es la que debe componer alrededor de 97% de la hemoglobina
adulta. La producción de HbS es el
resultado de la sustitución del aminoácido valina por el ácido glutámico en la
sexta posición de la cadena ϒ-globina. Cuando se expnen a una menor presión de
oxigeno los hematíes asumen la forma de una hoz, lo que causa su estasis a
nivel de los capilares.
MANIFESTACIONES CLINICAS.
·
Dolor musculoesquelético.
·
Esplenomegalia
·
Irritabilidad
·
Tumefacción dolorosa y aguda de manos y pies.
·
Dolor abdominal o visceral.
·
Nuemonía.
·
Síndrome torácico agudo
DIAGNÓSTICO.
La electroforesis de hemoglobina
confirma el diagnóstico y es útil para identificar variantes de hemoglobina como
la Hb fetal y la Hb A2.
En frotis de sangre periférica puede
revelar células falciformes, dianocitos, poiquilocitosis, hipocromía,
reticulocitosis, leucocitosis y trombocitosis.
También son comunes las
elevaciones de bilirrubina y LDH, puede existir elevaciones del BUN y la
cretinina en pacientes con insuficiencia renal progresiva.
El análisis de orina puede
revelar hematuria y proteinuria.
TRATAMIENTO.
Mantener una hidratación
adecuada. Corregir la hipoxia. Tratar de
manera agresiva las presuntas infecciones.
Proporcionar analgesia durante las crisis vasooclusivas. La hidroxiurea
500 a 700 mg/día eleva los niveles de hemoglobina F y reduce la incidencia de
complicaciones vasooclusivas.
ANEMIA NORMOCITICA NORMOCROMICA.

Generalmente son producidas por
hemólisis, hemorragias agudas, tumores malignos, esplenomegalia, agentes
tóxicos, enfermedades crónicas, artritis reumatoidea y enfermeades renales.
Manifestaciones clínicas.
DIAGNÓSTICO
El VCM es de 80 a 94 ft
HbCM 27 a 32 pg
CHbCM 32 a 36 g/
ANEMIA MICROCITICA HIPOCROMICA.
Se produce por un nivel de hierro
insuficiente para mantener la eritropoyesis normal y se caracteriza por
resultados anormales en los estudios del hierro.
MANIFESTACIONES CLINICAS.
palidez
taquicardia
atrofia de las papilas linguales
uñas frágiles
queilosis
estomatitis angular
pica y pagofagia.
DIAGNÓSTICO
VCM menor a 80 ft
CHbCM menor de 32 g/dl
En el extendido se observan
células pequeñas que tienen aumento de la palidez central.
Recuento de reticulocítos variable. los indices de producción de los mismos son bajos.
TRATAMIENTO
Sales de hierro orales (sulfato ferroso)
Bibliografía.
Ferri, Fred F., (2006). Consultor Clínico: Claves diagnósticas y
tratamiento. 1ª edición. España. Editorial Elsevier S.A.
Ruíz A, G.J., (2009). Fundamentos
de hematología. 4ª edición. México. Editorial Médica Panamericana.
https://books.google.com.gt/books?id=fH5IQrCxcskC&pg=PA300&dq=ANEMIA+HEMOL%C3%8DTICA+sintomatolog%C3%ADa&hl=es&sa=X&ved=0CBoQ6AEwAGoVChMIrsTI5LOUyQIVRu4mCh2zrAK2#v=onepage&q=ANEMIA%20HEMOL%C3%8DTICA%20sintomatolog%C3%ADa&f=false
 Linfoma No Hodking
Linfoma No Hodking Linfoma No Hodking
Linfoma No Hodking